
The Importance of K-mer Frequency in Genomic Analysis
A k-mer is a contiguous sequence of k nucleotides (the building blocks of DNA) in a genome. Biologists often use k-mers to identify patterns or motifs in genomic sequences, such as repeated sequences or conserved regions.
This can be useful for a variety of purposes, such as identifying potential regulatory elements or identifying DNA sequences that are conserved across different species.
In genomics, a conserved region refers to a sequence of nucleotides (the building blocks of DNA) that is highly similar or identical across different species or organisms. These conserved regions may be found within a genome at the DNA level, or they may be found at the level of the protein sequences that are encoded by the DNA. Conserved regions play a key role in understanding the evolution and function of different species and can provide valuable insights into the structure and function of genomic sequences.
rebelScience.club article: Genome Toolkit. Part 3: Building statistical data (k-mer frequency)
One example of where this might be useful is in the analysis of transcription factor binding sites. Transcription factors are proteins that bind to specific DNA sequences and regulate gene expression. By identifying the most frequent k-mers in a genome, biologists can identify potential binding sites for transcription factors and study how these sites are regulated in different cell types or under different conditions.
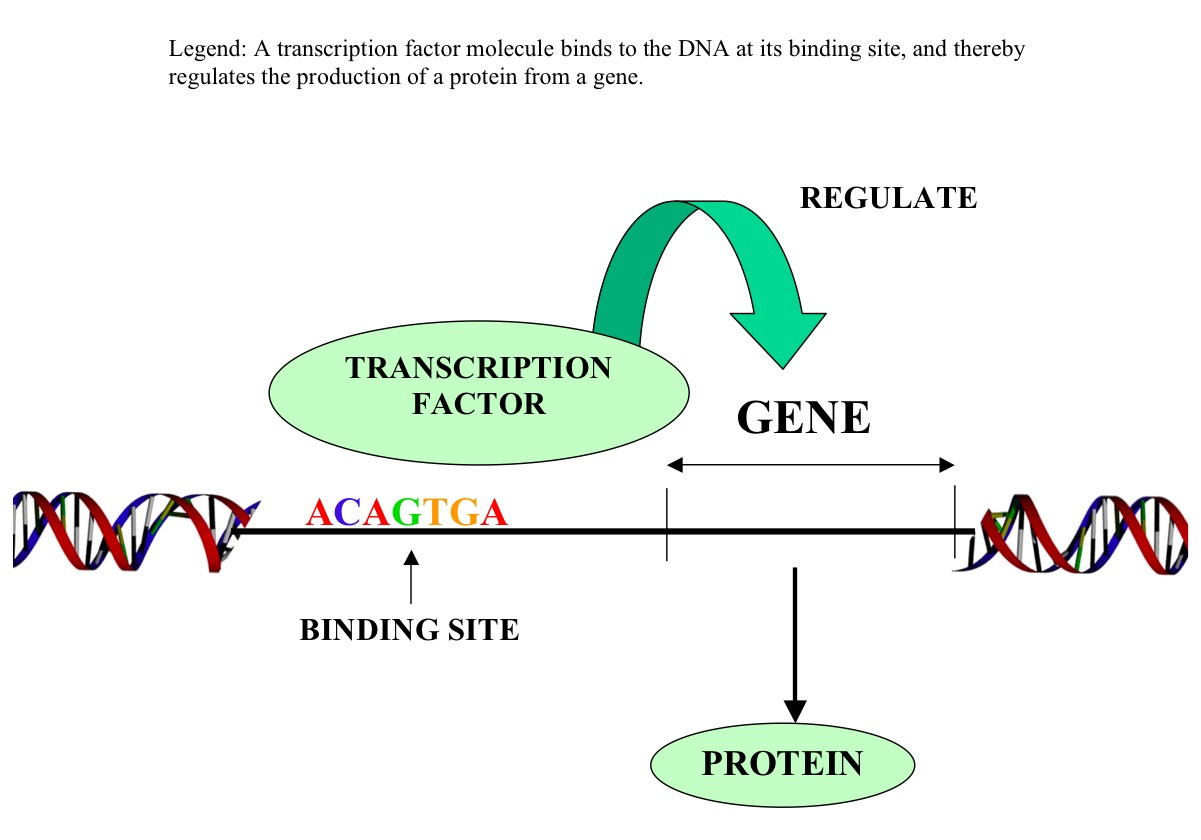
Biologists can use k-mer analysis to identify conserved sequences across different species. This can help them to understand the evolutionary relationships between different organisms and identify potentially functional sequences that have been conserved over time.
Let’s now add such an algorithm to our Genome Toolkit, and break it down.
The algorithm
First, I want to start by updating the way we will be writing comments and documentation for our algorithms. We will be using extended dockstrings. If you have completed the DNA Toolkit series, you should be familiar with dockstrings. Docstrings are comments written in triple quotes (''' or """) that provide a brief description of a function or code block. They can include information about parameters, return value, and any special considerations. Docstrings are useful for documenting your code and making it more readable and understandable for others. They can also be used to generate automated documentation.
We are going to update our first algorithm with the following documentation:
def count_kmer(self, sequence, kmer):
"""
Counts the number of times a specific k-mer appears in a given sequence,
including overlapping k-mers.
Parameters:
sequence (str): The DNA sequence to search in.
kmer (str): The specific k-mer to search for in the sequence.
Returns:
int: The number of times the k-mer appears in the sequence.
"""
kmer_count = 0
for position in range(len(sequence) - (len(kmer) - 1)):
if sequence[position:position + len(kmer)] == kmer:
kmer_count += 1
return kmer_countNow, let’s write our frequent kmers algorithm. There are a few ways of approaching this algorithm. We will use the dictionary approach, to avoid using any additional Python modules, like Counter. We will try using additional helper modules only when there is a significant benefit to the performance of our code.
Our algorithm will have four simple parts:
- A dictionary to store k-mer frequencies.
- A loop to iterate through the DNA string and extract k-mers of a given length, while also incrementing the frequency of each k-mer.
- A variable to store the maximum value in the dictionary
- List comprehension/loop to scan the k-mer dictionary, and return a list of k-mers with the highest frequency.
And here is the implementation:
def find_most_frequent_kmers(self, sequence, k_len):
"""
Finds the most frequent k-mers of a given length in a DNA string.
Parameters:
sequence (str): The DNA string to search.
k_len (int): The length of the k-mers to search for.
Returns:
list: A list of the most frequent k-mers in the DNA string.
"""
# 1. A dictionary to store k-mer frequencies.
kmer_frequencies = {}
# 2. A loop to iterate through the DNA string and extract k-mers of a given length,
# while also incrementing the frequency of each k-mer.
for i in range(len(sequence) - k_len + 1):
kmer = sequence[i:i+k_len]
if kmer in kmer_frequencies:
kmer_frequencies[kmer] += 1
else:
kmer_frequencies[kmer] = 1
# 3. A variable to store the maximum value in the dictionary
highest_frequency = max(kmer_frequencies.values())
# 4. List comprehension/loop to scan the k-mer dictionary and return a list of k-mers
# with the highest frequency.
return [
kmer for kmer, frequency in kmer_frequencies.items()
if frequency == highest_frequency
]This is a very simple, yet powerful function. In the last part of our code, we separate the return statement into 4 lines instead of one long line. It is easier to read that way, and it does not go off the screen.
Let’s also look at another way of writing this algorithm:
from collections import Counter
def find_most_frequent_kmers(self, seq, k_len):
kmer_list = [
seq[i:i+k_len] for i in range(len(seq) - k_len + 1)]
kmer_frequencies = Counter(kmer_list)
highest_frequency = max(kmer_frequencies.values())
return [
kmer for kmer, frequency in kmer_frequencies.items()
if frequency == highest_frequency
]This version of the function uses the Counter function to count the frequency of each k-mer in the DNA string, rather than a loop. This can be more concise and easier to read, but may be slightly slower than using a dictionary due to the overhead of creating a new Counter object.
We will use the first version in our Genome Toolkit class. It eliminates the unnecessary list comprehension and Counter function calls, and instead uses a loop and a dictionary to store and count the k-mer frequencies.
To summarize
K-mer frequency is useful for a variety of purposes in genomics and biology, including:
- Identifying potential regulatory elements within a genome: By analyzing the k-mer frequencies of different k-mers within a genome, biologists can identify repeating patterns and motifs that may be involved in the regulation of gene expression.
- Understanding the evolutionary relationships between different species: Comparative genomics involves the study of the genomic sequences of different species in order to understand their evolutionary relationships. K-mer frequency analysis can be used to identify conserved sequences across different species, which can help biologists to understand the functional importance of these sequences.
- Analyzing transcription factor binding sites and gene regulation: Transcription factors are proteins that bind to specific DNA sequences and regulate gene expression. By analyzing the k-mer frequencies of different k-mers within a genome, biologists can identify potential binding sites for transcription factors and study how these sites are regulated in different cell types or under different conditions.
- Identifying conserved sequences across different species: K-mer frequency analysis can be used to identify sequences that are conserved across different species, which can help biologists to understand the functional importance of these sequences and the evolutionary relationships between different organisms.
Overall, k-mer frequency analysis is a valuable tool for biologists looking to build statistical data and identify patterns and motifs within a genome. It has numerous applications in genomics and biology and can provide valuable insights into the structure and function of genomic sequences.
Conclusion
After using the k-mer frequency algorithm to identify the most frequent k-mer in a genome sequence, we can then use the k-mer count algorithm to search for that specific k-mer in other genome sequences. This can be useful for identifying common patterns or features in different genome sequences and can aid in various types of analysis and research.
Let’s take a look at a few visualizations of this process:
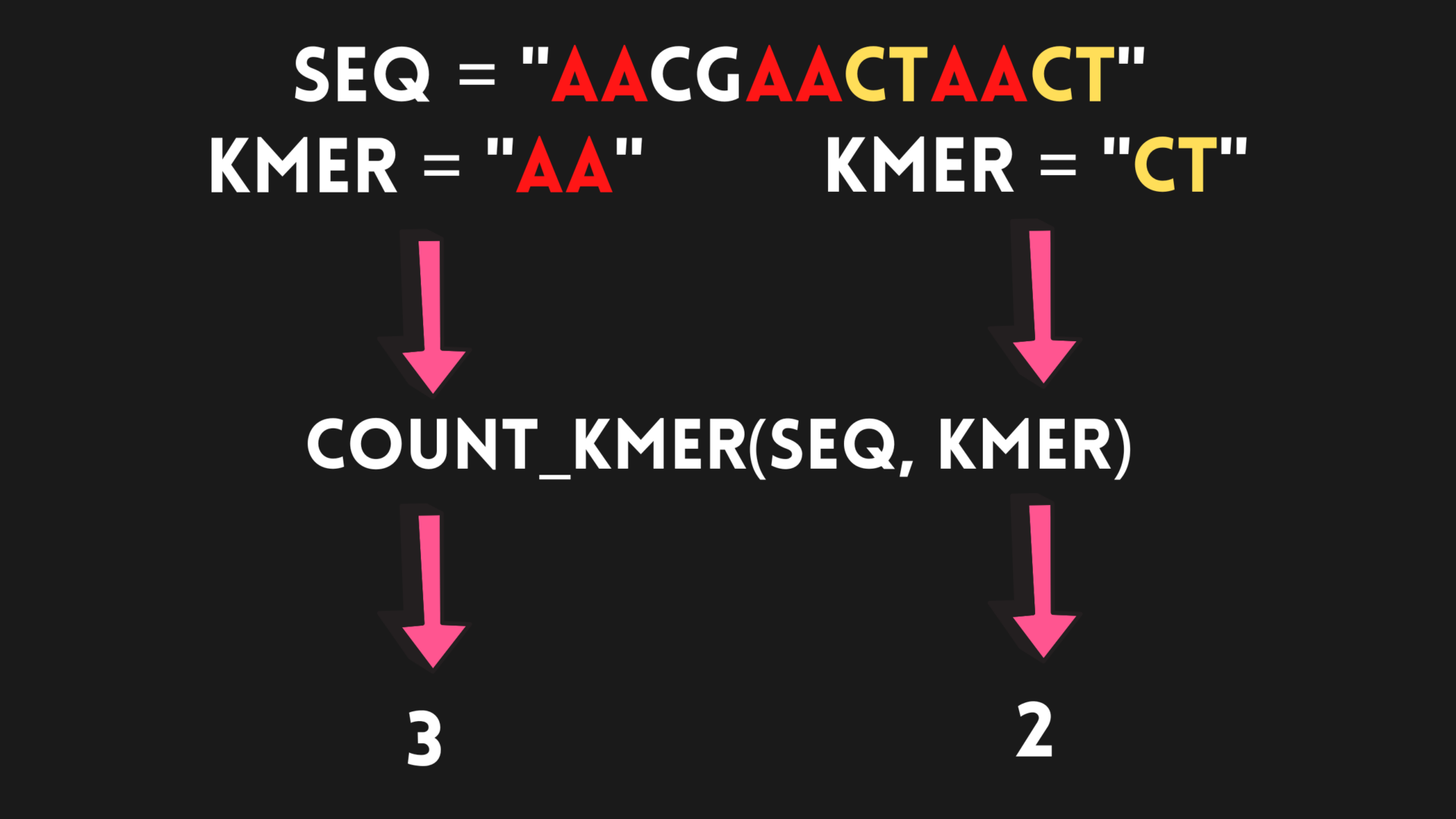




However, it’s important to note that this is still a naive approach, as it does not take into account mutations in DNA known as single nucleotide polymorphisms (SNPs). SNPs are variations in a single nucleotide (A, T, C, or G) at a specific position in a genome that can occur naturally and are quite common in the human genome. Since a given k-mer can appear in other genome sequences with a slight mutation due to an SNP, our algorithm may not be able to detect it.
In future articles, we will expand our algorithms to take these types of mutations into account in order to more accurately analyze and compare genome sequences. Overall, the use of k-mer frequency and k-mer count algorithms can be a useful starting point for analyzing genome sequences, but it’s important to consider the limitations of these approaches and continue to develop and refine our methods as needed.
Shoutout
A huge shoutout to Joyce Shi for the amazing PayPal donation and to Joshua for the amazing Patreon support! Your donations are truly appreciated and make a big difference in helping the rebelScience project grow and improve. Your support is motivating and much appreciated. Every dollar and every donation help the project grow, and I am grateful for your generosity. Thank you for your support!
If you found this video useful, please consider supporting the rebelScience project, and open education online, by buying me a coffee:

Genome Toolkit GitHub code repository can be found here: https://github.com/rebelC0der/Genome_Toolkit
Video version of this article:

