
In this article, we conclude our work on a minimal set of functions to work with DNA. We will add the last three functions that will help us to search for proteins in DNA sequences by generating reading frames. We will also apply our code to a real piece of genome that codes for Homo Sapiens Insult protein to see our code in action.
Functions we will add:
- Reading frame generation.
- Protein Search in a reading frame (sub-function for the next function).
- Protein search in all reading frames.
Let’s take a look at how codons (DNA nucleotides triplets) form a reading frame. We just scan a string of DNA, match every nucleotide triplet against a codon table, and in return, we get an amino acid. We keep accumulating amino acids to form an amino acid chain, also called a polypeptide chain. Here is a nice image that shows a reading frame:
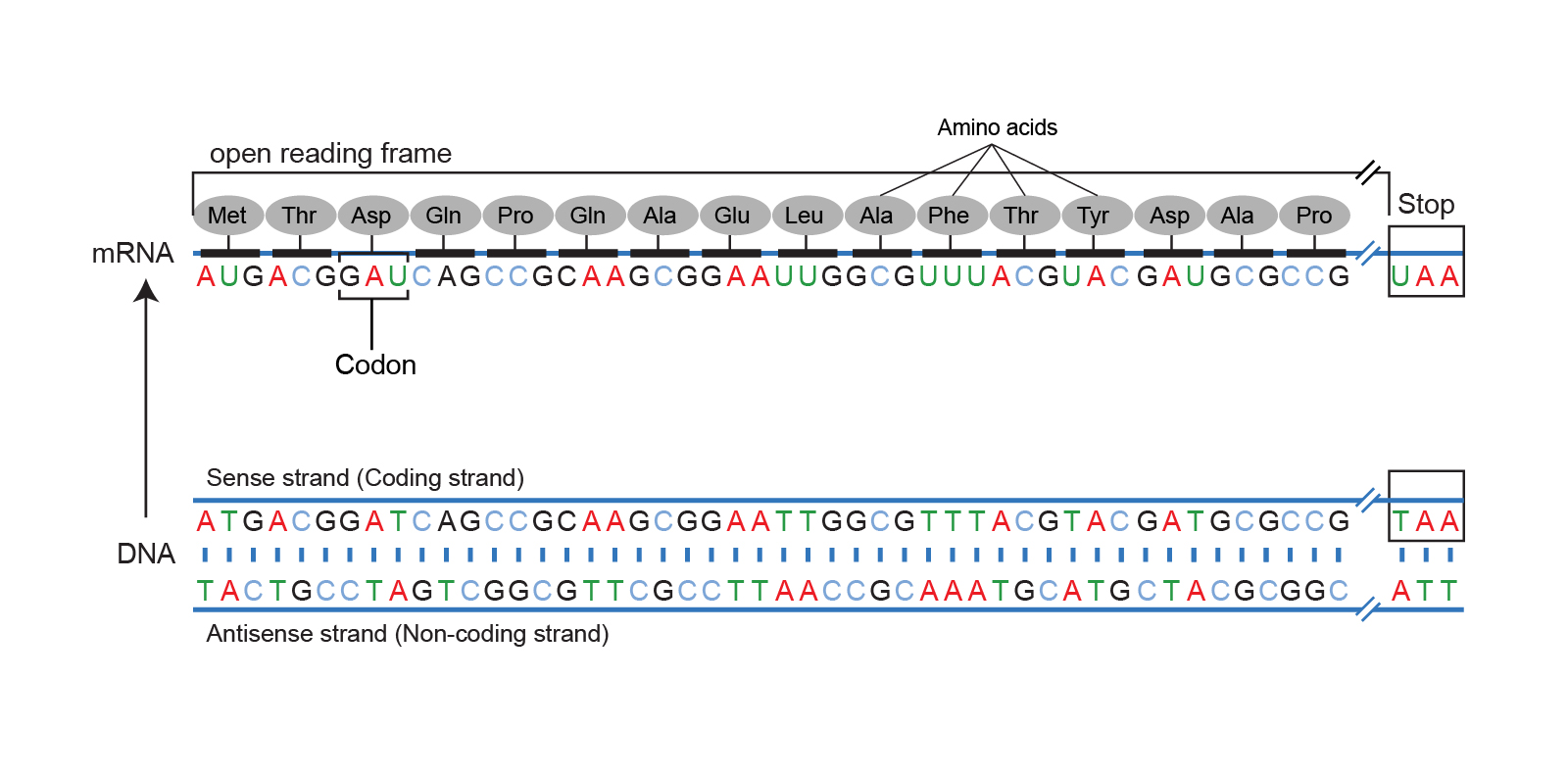
Also, here is a very nice explanation (audio) of what a reading frame is and why we need to form six of them for a proper protein search:
So let’s implement our reading frame generator to replicate the biological process that is performed by Ribisome in a living cell. We will reuse translation and reverse complement functions from our previous articles. In biology, Ribosome is an incredibly complex machine, but in the code, it is very simple:
def gen_reading_frames(seq):
"""Generate the six reading frames of a DNA sequence"""
"""including reverse complement"""
frames = []
frames.append(translate_seq(seq, 0))
frames.append(translate_seq(seq, 1))
frames.append(translate_seq(seq, 2))
frames.append(translate_seq(reverse_complement(seq), 0))
frames.append(translate_seq(reverse_complement(seq), 1))
frames.append(translate_seq(reverse_complement(seq), 2))
return frames- We create a list to hold lists of amino acids. This list will hold six lists.
- We add the first three reading frames, 5′ to 3′ end, by shifting one nucleotide in each frame. Our translation function accepts a string as a first argument and a start reading position as a second argument.
- We do the same operation three more times, but we generate the reverse complement of our sequence first.
We can now add a 9th output to our main.py file. It is a loop in this case, as we need to print 6 lists.
print('[9] + Reading_frames:')
for frames in gen_reading_frames(DNAStr):
print(frames)If we now run this function on the string below, here is what we should see:
GGGCGGCTCG ['G', 'R', 'L'] ['G', 'G', 'S'] ['A', 'A'] ['R', 'A', 'A'] ['E', 'P', 'P'] ['S', 'R']
The next function looks very convoluted, but it is very simple in principle. It uses an amino acid list as an argument and scans it to see if it contains a START – M codon and a STOP – _ codon. When M codon is found (lines 13 – 16) we start accumulating every amino acid after that, until will come across a _ codon (lines 7 – 11). We have two lists here: current_prot[] holds a current protein, being accumulated and proteins[] holds all found proteins in a sequence. This is needed because an amino acid sequence may contain multiple START – STOP codons, resulting in multiple possible proteins in a single sequence. Using a debugger to scan this function line by line will help to understand this code better. I also cover debugging this function in my video version of this article here.
def proteins_from_rf(aa_seq):
"""Compute all possible proteins in an aminoacid"""
"""seq and return a list of possible proteins"""
current_prot = []
proteins = []
for aa in aa_seq:
if aa == "_":
# STOP accumulating amino acids if _ - STOP was found
if current_prot:
for p in current_prot:
proteins.append(p)
current_prot = []
else:
# START accumulating amino acids if M - START was found
if aa == "M":
current_prot.append("")
for i in range(len(current_prot)):
current_prot[i] += aa
return proteinsIn this case, we are not adding an output as this function is a sub-function for our next function, and it only builds a protein from a single reading frame. Our next function will use this code and pass all six reading frames to it.
We can still do a quick test:
print(proteins_from_rf(['I', 'M', 'T', 'H', 'T',
'Q', 'G', 'N', 'V', 'A', 'Y', 'I', '_']))And this protein sequence will be generated: ['MTHTQGNVAYI']
Let’s now add our final function, that is a part of a pipeline. This function uses a few previous functions to generate a list of proteins for us, and it accepts 4 arguments; a sequence, a start reading and stop reading positions, and a boolean flag that lets us sort the list from a longest to a shortest protein sequence.
def all_proteins_from_orfs(seq, startReadPos=0, endReadPos=0, ordered=False):
"""Compute all possible proteins for all open reading frames"""
"""Protine Search DB: https://www.ncbi.nlm.nih.gov/nuccore/NM_001185097.2"""
"""API can be used to pull protein info"""
if endReadPos > startReadPos:
rfs = gen_reading_frames(seq[startReadPos: endReadPos])
else:
rfs = gen_reading_frames(seq)
res = []
for rf in rfs:
prots = proteins_from_rf(rf)
for p in prots:
res.append(p)
if ordered:
return sorted(res, key=len, reverse=True)
return resWe start by checking if the reading position was provided (lines 5 – 8). If yes, we generate reading frames for a slice of the string, if not, we generate reading frames for the whole sequence. This allows us to pass a sequence and just specify (if needed) a slice of it to look for proteins in, instead of pre-formatting (slicing) a string before providing it to our function.
Lines 10 – 14 we scan all six reading frames and using our previous function to generate all possible proteins in a reading frame. We end up with a list res[] that contains all protein sequences, found in all six reading frames.
I mentioned a pipeline above, as this function uses previous functions to produce the result. We start with a DNA sequence and we have this pipeline by using this function:
DNA -> Translation -> Reverse Complement -> Reading Frame Generation -> Protein Assembly
Now let’s add our final, 10th output loop:
print('\n[10] + All prots in 6 open reading frames:')
for prot in all_proteins_from_orfs(DNAStr, 0, 0, True):
print(f'{prot}')The best way to test out the final function is to apply it to a real biological sequence. Homo sapiens insulin, variant 1. It can be found on NCBI database. If we look at a FASTA formatted file, we see this DNA sequence:
https://www.ncbi.nlm.nih.gov/nuccore/NM_001185098.1?report=fasta
Let’s add it to our sequences.py file:
# NM_000207.3 Homo sapiens insulin (INS), transcript variant 1, mRNA NM_000207_3 = '\ AGCCCTCCAGGACAGGCTGCATCAGAAGAGGCCATCAAGCAGATCACTGTCCTTCTGCCAT\ GGCCCTGTGGATGCGCCTCCTGCCCCTGCTGGCGCTGCTGGCCCTCTGGGGACCTGACCCA\ GCCGCAGCCTTTGTGAACCAACACCTGTGCGGCTCACACCTGGTGGAAGCTCTCTACCTAG\ TGTGCGGGGAACGAGGCTTCTTCTACACACCCAAGACCCGCCGGGAGGCAGAGGACCTGCA\ GGTGGGGCAGGTGGAGCTGGGCGGGGGCCCTGGTGCAGGCAGCCTGCAGCCCTTGGCCCTG\ GAGGGGTCCCTGCAGAAGCGTGGCATTGTGGAACAATGCTGTACCAGCATCTGCTCCCTCT\ ACCAGCTGGAGAACTACTGCAACTAGACGCAGCCCGCAGGCAGCCCCACACCCGCCGCCTC\ CTGCACCGAGAGAGATGGAATAAAGCCCTTGAACCAGC'
The main page shows a protein sequence we should get from that DNA sequence:
https://www.ncbi.nlm.nih.gov/nuccore/NM_001185098.1
/translation="MALWMRLLPLLALLALWGPDPAAAFVNQHLCGSHLVEALYLVCGERGFFYTPKTRREAEDLQVGQVELGGGPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCN"
So let’s run our code and see if we can generate this protein:
print('\n[10] + All prots in 6 open reading frames:')
for prot in all_proteins_from_orfs(NM_000207_3, 0, 0, True):
print(f'{prot}')And here is what we see:
[10] + All prots in 6 open reading frames: MALWMRLLPLLALLALWGPDPAAAFVNQHLCGSHLVEALYLVCGERGFFYTPKTRREAEDLQVGQVELGGGPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCN MRLLPLLALLALWGPDPAAAFVNQHLCGSHLVEALYLVCGERGFFYTPKTRREAEDLQVGQVELGGGPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCN MLVQHCSTMPRFCRDPSRAKGCRLPAPGPPPSSTCPTCRSSASRRVLGV MPRFCRDPSRAKGCRLPAPGPPPSSTCPTCRSSASRRVLGV MAEGQ ME
We can see that the first protein in our output indeed matches NCBI proposed protein, confirming the correctness of our code in every step of the pipeline.
Alright! This is it. Now we have a set of basic tools to work with DNA. Next, we will refactor this code into a nice reusable class and optimize some code. This might be overkill for an article, so if you are interested in wrapping this code into a class, feel free to watch parts 8 to 9, as they come out, here:
GitHub repository: https://github.com/rebelC0der/DNA_Toolkit
Until next time, rebelCoder, signing out.
Why are we translating a DNA strand into a protein ? I thought we should first take the DNA strand, transcribe it to its RNA form, and then generate open reading frames on the RNA strand. Then take extract protein information from this orfs?